.svg)
LLMS are not the hard part: Considerations for AI-Ready Data

By:
,
Viraj Narayanan
CEO
of Cornerstone AI
My journey at Cornerstone AI is built on my prior experiences watching life sciences companies grapple with the same problem from different angles. At COTA, we worked on getting unstructured oncology data out of EMRs to inform drug research. At Ontada, we ran an EMR for community oncology and used those data to power biopharma research and commercialization. In both roles I noticed that the science was ready to move faster than the data underneath it could support, and the gap between those two speeds was that infrastructure repeatedly slowed down promising ideas. This dynamic helped me understand why people would say that biopharma can be a “data rich, but insights poor” environment.
That pattern is now the central conversation in biopharma, except the stakes have changed. Every executive I meet has the same vision, which is that every life sciences company will have its own LLMs powering faster and more comprehensive decisions across clinical development, with trials designed in days rather than months, protocols optimized against thousands of historical comparators, and patients identified as candidates before a coordinator opens a chart. I believe in that vision and am committed to working toward it every day. However, there remains a gap between the boardroom and the data scientist’s desk that we should be honest about. Biopharma executives have a paper map that shows how their teams will get to the target destination, while the technical stakeholders on the ground are using Waze or Google Maps, which show every bump in the road, traffic jam, road work, and speed trap, highlighting that the journey towards AI optimization is much more difficult to traverse than the AI conversation has acknowledged. The executive's paper map is not wrong; it simply cannot see the live conditions that decide whether you arrive on time. The data team is the only one watching real-time traffic, and that is precisely why the executive and the desk-level reality keep drifting apart.
AI-ready: more than data quality
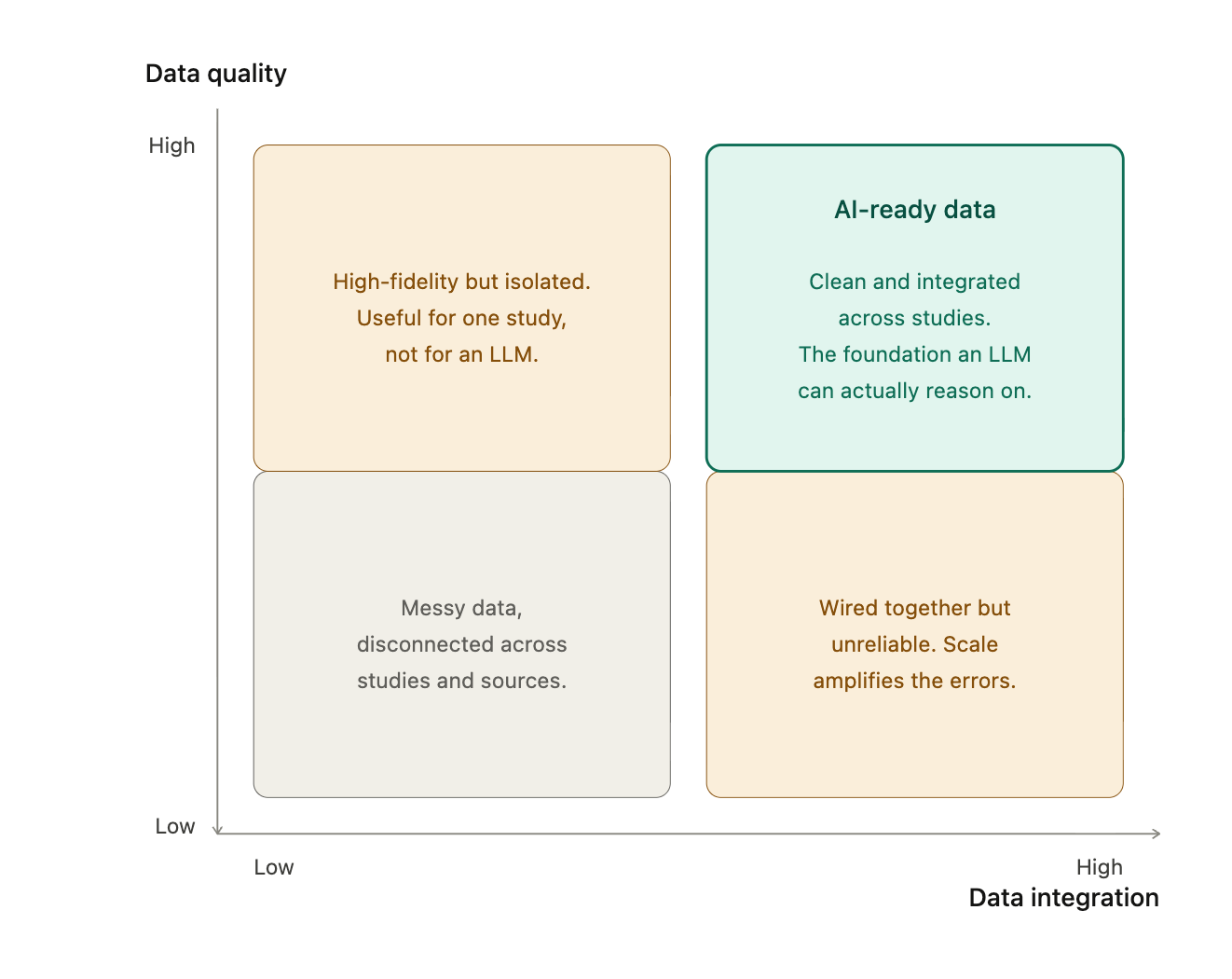
When people say "AI-ready,” they usually refer to “clean data,” which is roughly half of what the term should mean. The other half is about integration, and integration is where most of the actual work lives. A single trial sits across ten to fifteen tables that come from a variety of sites, sometimes with distinct principal investigators. A major sponsor runs three hundred trials in parallel, and that same sponsor is sitting on thousands of historical ones (a top 50 pharma has between 2,000 to 4,000 historical trials). To power an LLM that can actually reason across a therapeutic area, you don't just need each dataset to be clean, you must ensure that they can talk to each other, which means the same data model, the same vocabulary, and the same definitions of an adverse event or a dose modification or a screen failure, across studies that were each originally designed to answer a different scientific question. To make that concrete: the same adverse event coded three different ways across three studies will silently corrupt any cross-trial reasoning built on top of it, and nothing in the output will announce that it happened. Each trial is its own world by design, because the protocol authors made deliberate choices to answer a specific question, and the adverse event coding decisions, lab panel selections, and derived variable definitions all carry the fingerprint of that one trial's purpose. That's good science, and it's also why pooling trials for an LLM is not a copy-paste exercise.
A 2024 Komodo Health report put a number on this that has stuck with me, which is that roughly 70% of life sciences teams invest six or more months transforming data into a usable state before they can even start to derive insights, and roughly half of those teams rely on four or more consultants to do it. That was the state of play before LLMs raised the bar - and the bar is much higher today.
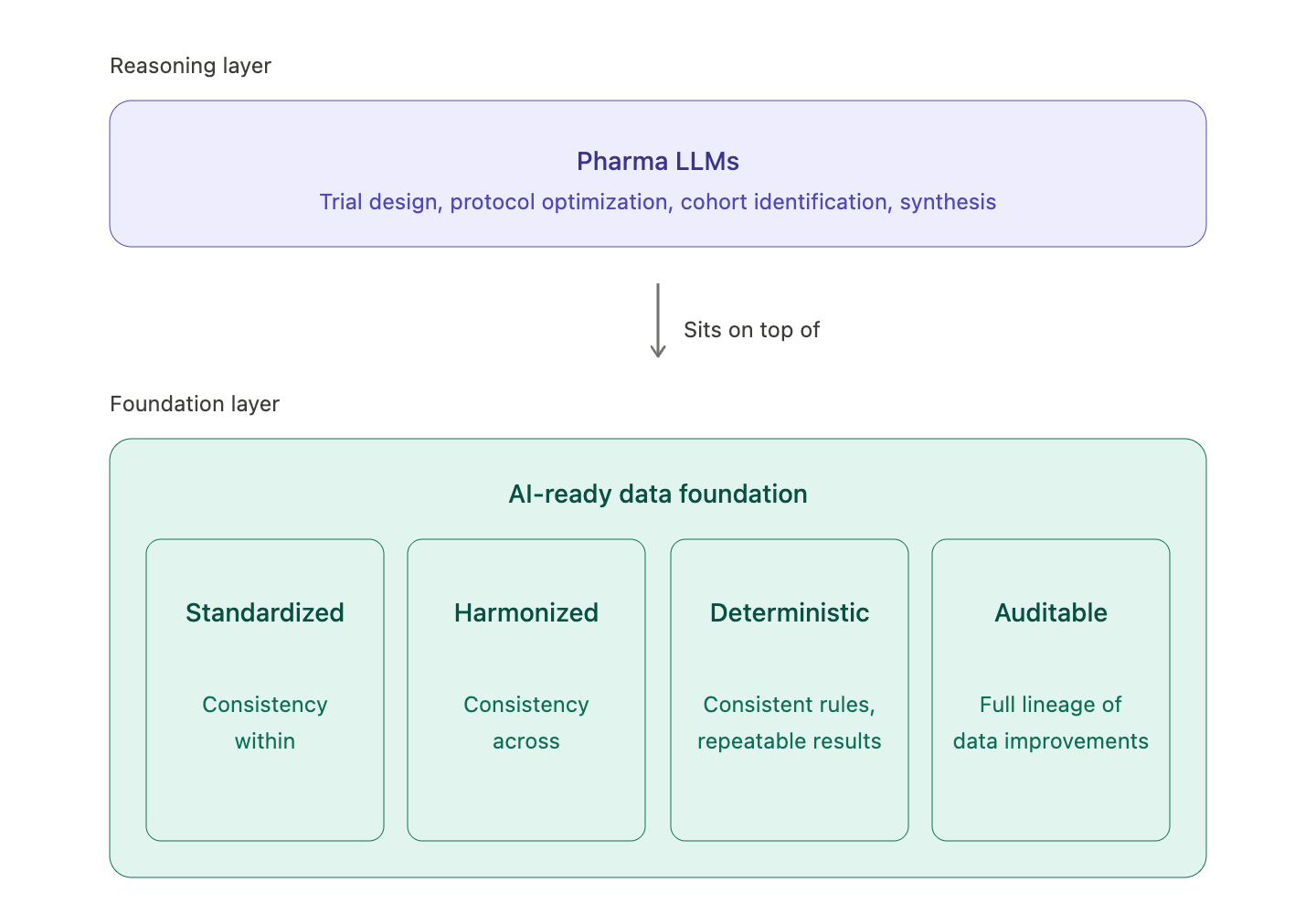
Where LLMs actually fit
The conversation I'd like to see more of in our industry is about where LLMs belong in the stack; effectively, you can't solve for the LLM before solving for the data foundation, or else you're wasting time. My view, which has sharpened over the past year of customer work, is that LLMs sit on top of the data foundation, not inside it where it will produce a different foundation each time it is run. The foundation must be deterministic, versioned, reproducible, traceable, and auditable, because a submission package needs to show that the same input produces the same output today as well as three years from now when an inspector asks. LLMs are a different kind of tool, brilliant at reasoning, synthesis, and surfacing patterns that humans miss but only within the box of context they have. They’ll brilliantly reason wrong outputs if trained on messy or incorrect inputs, which is why they earn their true value when they sit on top of a foundation that we can trust. In other words, the basic requirements of the regulatory process have not changed just because AI and LLMs have come into the picture.
There's a second reason why this layering matters, which is that pharma clinical trial data is core intellectual property. Each sponsor has spent decades and billions of dollars generating it, and the comparative knowledge buried inside it, like how patients responded across arms, how endpoints behaved across populations, and how safety signals evolved over time, is among the most valuable IP a sponsor owns. That data is not going to be aggregated across sponsors at the model layer, and it shouldn't be. Which means that biopharma will soon come to appreciate that future LLMs aren’t a single shared general-purpose model that has somehow absorbed the industry's knowledge; they are each sponsor's own models, reflective of a sponsor’s reasoning and judgment and manifest in a sponsor’s own data, with the foundation underneath built to a standard that the model can rely on and this is where we have been focused. The stakes here are best illustrated by what happens when a model is pointed directly at unverified data: it hallucinates, and in this domain the hallucinations are uniquely dangerous because they are fluent and confident. A fabricated safety signal or a wrong dose-modification summary reads exactly like a correct one, with no obvious tell. Because the failure is invisible at that moment, it surfaces downstream, after a go/no-go call on a billion-dollar program has already been made on the strength of an answer that was never true.
What we've learned doing the work
We have been privileged to work with some of the largest life sciences companies in the world on this problem, and a few patterns are clear from our work.
- The first is that speed is the headline but accuracy is the contract. When we cut a sponsor's trial harmonization timeline from months to days, the case study writes itself, but what doesn't show up in the case study is the additional weeks the sponsor spent reviewing record by record before they trusted the output. They were right to do that work, because if you're going to feed an LLM that informs a go/no-go decision on a billion-dollar program, one bad data point is not an acceptable error rate. The bar is total confidence, and earning it requires traceability that survives audit. This is a lesson I first encountered at COTA in the RWE conversations for regulatory purposes, and it has only gotten more important as the downstream consumer of the data has become a model rather than a human reviewer.
- The second pattern is that sponsors must treat this type of work as core infrastructure versus a one-time initiative. Every new trial, every new data source, and every new schema requirement trigger a process to ensure the underlying data is accurate, consistent and interoperable. The companies that will pull ahead are the ones that treat it as infrastructure, the way they treat their cloud or their EDC or their safety database, because the alternative is an endless cycle of consultants and rework that the Komodo report described.
- The third pattern is the one I think about the most, which is that the patient is on the other side of all of this. A faster trial is a faster therapy, and a more comprehensive cohort is a patient who gets identified as a candidate when they otherwise would not have been. Because the average drug takes ten to fifteen years and 2.5 billion dollars to get approved, every month spent wrestling data into shape is a month a patient with a narrowing set of options spends waiting for an answer, and many of them do not have that kind of time.

The work ahead
The pharma LLM era is coming, and the question is not whether sponsors will deploy these models, it’s what the models will be reasoning over. If the answer is the data as it sits today, the era will disappoint, and if the answer is data that is genuinely AI-ready, meaning clean and integrated and built on a foundation the model can trust, then the vision executives describe is not a five-year fantasy but a near-term operating reality. That is the unlock, and it's why we are heads down on the work. If any of the above resonates with what you are seeing in your own organization, I'd love to compare notes.
My thanks to Chris Boone, Dan Jacob, Vera Mucaj, Marisa Co, and Jeremy Brody for their comments on earlier iterations of this article.
[i] https://www.fda.gov/media/75414/download
[ii] https://pmc.ncbi.nlm.nih.gov/articles/PMC12318031/
.jpg)