.svg)
What Accelerated Approval Can Teach the FDA's Real-Time Clinical Trials Pilot

January, 2000. Mike during his FDA tenure
By:
,
Mike Elashoff
CSO, Co-Founder
of Cornerstone AI
The FDA recently opened a request for information on a pilot to use AI to enable faster early-phase trial decisions. I submitted a comment, and I want to share why I think the agency is onto something important.
There are clear parallels to an earlier FDA advance: accelerated approval. I was a reviewer in the Antiviral division during the height of the HIV/AIDS epidemic, at ground zero of that change. Accelerated approval let the FDA make faster decisions on drugs and delivered better outcomes for millions of patients. Critically, it did this without abandoning the standards of safety and efficacy that clinical trials exist to uphold. The current AI pilot has the same shape: reach faster decisions while holding rigorous standards in place. That is the right instinct, and it is why I am genuinely glad to see it.
I co-founded Cornerstone AI to bring AI to clinical trial data within those standards, not around them. So I have a few specific recommendations, which I submitted to the docket.
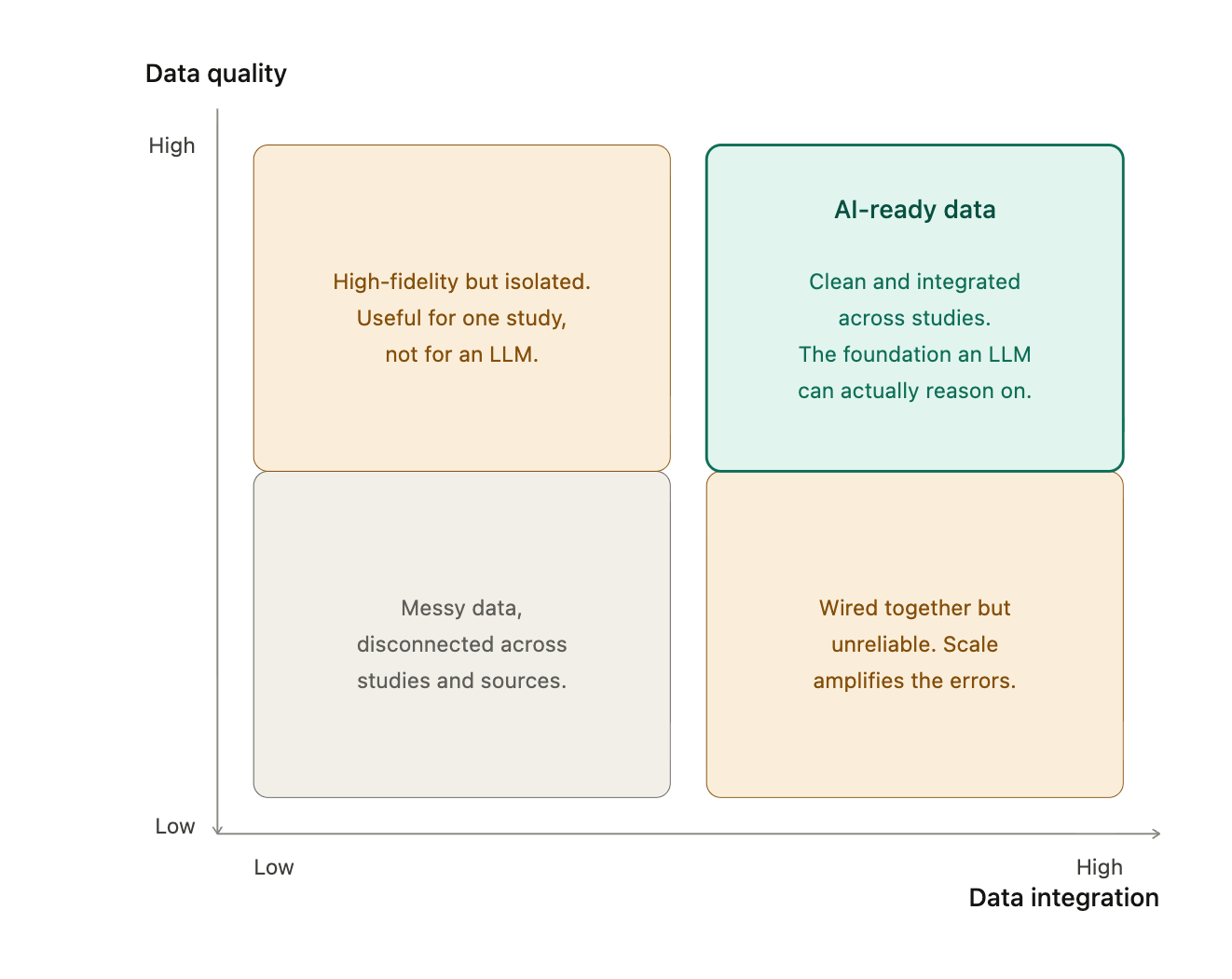
First, control data. Every clinical trial is, at its core, trying to answer one question: how does this drug compare to alternatives? Early trials make that hard. Small sample sizes and short follow-up are particularly unforgiving for rare or serious adverse events. One promising approach is using indication-specific, cross-trial harmonized datasets as a source of control data. In our work with pharma companies, we see them assembling exactly these datasets internally right now. The FDA could encourage harmonized datasets as part of early trial submissions to improve the power and accuracy of comparisons.
The catch is that harmonization is genuinely hard, because study-to-study differences exist even within the same drug program at the same sponsor. A real example from work we have seen: the same transaminitis adverse event was coded as "transaminitis" in one study's SDTM submission, as "hypertransaminasaemia" in a second, and as "transaminases increased" in a third. That heterogeneity runs across many events and quietly corrupts comparative analysis. Lab data is another common source of variance, given inconsistent standardization to controlled terminologies like LOINC. The good news is that AI can solve these problems when applied properly.
Second, on quality. We recommended the FDA include AI quality checks alongside early trial submissions. For context on why this matters: we see clinically significant errors in roughly 10 to 20 percent of EHR records, about 5 percent of pre-datalock EDC records, and under 0.5 percent of post-datalock EDC records. To really get the benefit of live trial decision making, the traditional datalock procedure has to be near real time as well.
Third, on trust. We recommended the FDA require the AI models used for live trials be fully reproducible and fully auditable. When a regulator needs to understand why a data point was changed, "the model decided" cannot be the answer. We have built 21 CFR Part 11 validated AI models precisely so that the promise of AI can operate inside a rigorous regulatory framework rather than asking the framework to bend.
In clinical research faster only matters if it is also trustworthy. The FDA asking how to get there is a promising start. We'd welcome a discussion as the agency considers how to build rigorous data quality approaches into this pilot.
You can read the full RFI and public comments here: https://www.regulations.gov/comment/FDA-2026-N-4390-0054
.jpg)